
Peripartum cardiomyopathy (PPCM) is a rare but potentially life-threatening condition that presents as heart failure (HF) during the peripartum period and may progress to cardiogenic shock. (1) Recent genetic studies indicate that up to 20% of PPCM patients carry mutations in genes associated with dilated cardiomyopathy (DCM), such as the TTN gene, which encodes titin, emphasizing the value of genetic testing in this population. (2) We describe a challenging case of PPCM with a severe clinical course and a novel truncating TTN variant, focusing on diagnostic complexity and management, particularly regarding implantable cardioverter-defibrillator (ICD) timing.
A 33-year-old Caucasian woman with no relevant medical history presented to the emergency department five days postpartum with sudden dyspnea, orthopnea, and chest discomfort. She had one previous uneventful full-term pregnancy and a miscarriage. Family history was negative for cardiac disease.
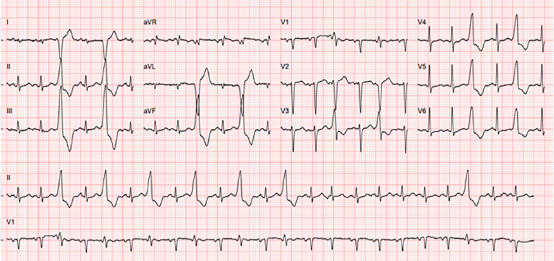
On admission, she was tachycardic, tachypneic, and hypotensive. Auscultation revealed an irregular rhythm, apical systolic murmur, and bilateral basal crackles. Laboratory testing showed elevated NTproBNP (N-terminal pro-B-type natriuretic peptide, 4400 pg./mL, normal value <450 pg./mL) and D-dimers (6000 ng/mL, normal value <500 ng/mL). In the computed tomography pulmonary angiography identified small bilateral pleural effusions. Electrocardiogram showed sinus tachycardia (140 bpm) with frequent ventricular bigeminy; QT interval was normal (Figure 1).
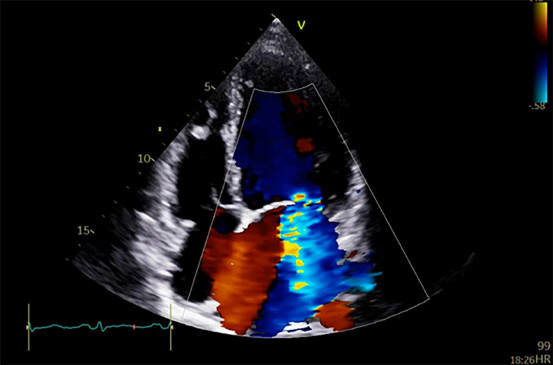
Echocardiography revealed a severely dilated and dysfunctional left ventricle (LV) with left ventricular ejection fraction (LVEF) 20%, borderline right ventricular (RV) function, and severe secondary mitral regurgitation (Figure 2).
Fig. 2
Transthoracic echocardiogram showing severe dilated left ventricle and severe secondary mitral regurgitation
She was admitted to the cardiac intensive care unit for clinical stabilization.
Within 24 hours, she deteriorated to cardiogenic shock and required levosimendan and norepinephrine. Lactation was suppressed using bromocriptine following shared decision-making with the patient. Anticipating need for mechanical support, she was transferred to a tertiary care center, where she responded to inotropic therapy with dobutamine without requiring circulatory assistance.
Disease-modifying drugs for heart failure were gradually introduced, though limited by her hemodynamic profile.
On day 23, cardiac magnetic resonance imaging (MRI) confirmed severe LV dilation and dysfunction (LVEF 28%) and mild RV dysfunction, without inflammation, fibrosis, or myocardial infiltration. She was discharged on day 40 and referred for genetic evaluation.
Follow-up in HF clinic allowed gradual uptitration of disease-modifying therapy. Genetic testing identified a novel, likely pathogenic, truncating variant in TTN (c.98319dup p.(Asp32774*)). First-degree relatives showed no abnormalities on echocardiography.
After 6 months of optimized therapy, cardiac MRI still showed severe LV dysfunction and a subcutaneous ICD was implanted. She ultimately achieved complete recovery of LV function (LVEF 54%) eight months post-admission.
Recent advances have provided new insights regarding the genetic basis of PPCM showing that up to 20% of patients with PPCM carry mutations in genes known to be associated to DCM. (2)
This case supports emerging data on the genetic underpinnings of PPCM, particularly the role of TTN truncating variants, which are known to correlate with adverse LV remodeling and reduced recovery.(2) The hypothesis of pregnancy as a physiological "second hit" that unmasks latent genetic DCM is reinforced. (3)
Data have shown that complete recovery of ventricular function occurs in a substantial proportion of women, and predictive factors include a baseline LVEF greater than 30%, as suggested by previous studies. (4) Although LVEF recovery can occur in up to 65% of patients by six months, (5) some recover only after a year or more. This variability complicates the timing of cardioverter-defibrillator implantation. Early permanent implantation may be premature, particularly in reversible cardiomyopathies, but data on ventricular arrhythmias in PPCM remain limited.
Some studies report high arrhythmic burden early in PPCM, with life-threatening ventricular arrhythmias observed in 12-43% of patients. (6) Wearable cardioverter-defibrillators (WCDs) have shown utility in this context. In one prospective series, 3 of 7 patients experienced ventricular fibrillation successfully terminated by the WCD. (6)
Although current guidelines are non-specific, WCDs may be a reasonable bridge strategy in patients with new-onset PPCM and severely reduced LVEF.
However, their limited availability in some countries may restrict routine use in clinical practice and complicate the decision of when to indicate an ICD.
This case illustrates the complexity of managing severe PPCM in the presence of a pathogenic TTN variant. It highlights the importance of genetic testing, the need for shared decision-making regarding lactation suppression, and the dilemma of ICD timing-particularly when an underlying genetic variant is associated with poorer left ventricular reverse remodeling.
Conflicts of interest
None declared.
(See conflicts of interest forms on the website).
Ethical considerations
Not applicable
Funding
No funding