Federico Sollazzo 1, Francisco C. Nutinez 1, Martín I. Maidana 1, Constanza B. Zacarías 1, Elián F. Giordanino 1 MTSAC, María F. Renedo1 MTSAC
1 Cardiology Service and Residencia. Hospital Universitario Fundación Favaloro
Address for correspondence: Federico Sollazo. E-mail: fedsollazzo@gmail.com
Rev Argent Cardiol 2024;92:157-159. http://dx.doi.org/10.7775/rac.v92.i2.20761
Arrhythmogenic cardiomyopathy (ACM) is a genetic disease of the cardiac muscle characterized by the progressive substitution of muscle cells by fibrous and adipose tissue, generally affecting the right ventricle (RV), with variable involvement of the left ventricle (LV), which causes ventricular dysfunction and predisposition to potentially fatal arrhythmias and sudden death. It is associated with dominant autosomal inheritance and irregular penetrance, involving genes encoding desmosomal proteins. Both its diagnosis and clinical management represent a challenge. (1)
This is the case of a 27-year-old male patient, with history of frequent ventricular extrasystoles (VES) in 2018, electrocardiogram with negative T waves in leads III, aVF, V2-V6, observed in a presurgical study. ECG-Holter monitoring evidenced 17 183 VES, bigeminies and triplets, during the day. The echocardiogram showed moderate left ventricular systolic function impairment, ejection fraction 40%, global hypokinesia, dilation and marked RV dysfunction, and cardiac magnetic resonance with inferior subepicardial, free wall and right ventricular outflow tract delayed enhancement, moderate left ventricular impairment and interventricular dyssynchrony, compatible with arrhythmogenic cardiomyopathy with biventricular involvement. These findings led to implantable cardioverter defibrillator (ICD) insertion as primary prevention of sudden death. Subsequently he was hospitalized in another institution for effective ICD shock, with evidence of multiple episodes of ventricular tachycardia (VT), and treated with betablocker and antiarrhythmic agents, sotalol at maximum doses and amiodarone, which had to be suspended for hyperthyroidism.
The patient consulted at our institution for frequent palpitations followed by ICD shock on two occasions. On admission, he was lucid, clinically stable, well perfused, without signs of congestion, no signs of pulmonary congestion, oxygen saturation at ambient air 98%, and no alterations in laboratory tests. Monitoring revealed repeated palpitations and sustained VT. (Figure 1) Due to confirmed ventricular arrhythmia and two ICD shocks in the last 24 hours, an electrical storm was diagnosed and a protocol with lidocaine and magnesium sulphate was initiated, with reversion to sinus rhythm.
The reading of ICD records was performed, and multiple episodes of sustained VT were ratified, with effective electrical cardioversion on two occasions and antitachycardia therapies on repeated circumstances.
A computed tomography angiography was performed for possible ischemic trigger, which ruled out coronary heart disease. Jointly with Electrophysiology, the possibility of radiofrequency ablation was evaluated by cardiac magnetic resonance, which confirmed right ventricular dilation with markedly increased volumes, severe systolic function impairment, and transmural fibrosis and thinning of the entire free wall. Severe thinning precluded ablation due to the high risk of ventricular rupture and cardiac tamponade.
The patient evolved with multiple episodes of electrical storm and resumption of the lidocaine protocol. Due to refractory response, orotracheal intubation, mechanical respiratory support and deep sedation was decided to decrease the adrenergic stimulus. He presented an episode of ventricular fibrillation, with cardiopulmonary resuscitation and external cardiac defibrillation that returned spontaneous circulation, followed by low cardiac output syndrome requiring the initiation of inotropic support with milrinone and levosimendan.
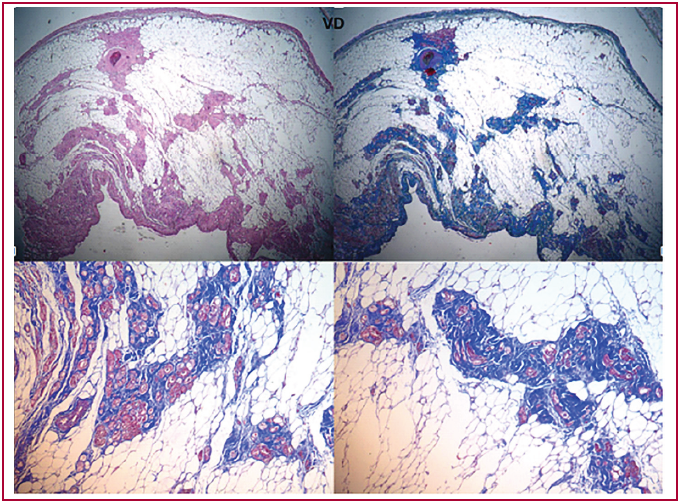
Because of his torpid evolution, the patient was evaluated for heart transplantation and subsequent incorporation into the waiting list in Emergency B condition. Mexiletine was initiated, which resolved a large proportion of the arrhythmic density, achieving extubation. Orthotopic bicaval heart transplantation was performed, without immediate postoperative complications. The native heart was sent for anatomo-pathological study, which established extensive adipose substitution and muscle bundles disorganization, with foci of myocyte necrosis, and final diagnosis of ACM with biventricular involvement. (Figure 2)
An endomyocardial biopsy was performed prior to discharge. It presented grade 2R cellular rejection with no evidence of antibody-mediated rejection, and was treated with high doses of corticoids with clinical and histopathological resolution. He subsequently completed his hospitalization uneventfully.
Arrhythmogenic cardiomyopathy is a rare hereditary heart disease, potentially fatal, that requires clinical care and a comprehensive therapeutic approach. The current diagnosis is based on the 2010 Task Force criteria, consisting of structural disorders in imaging and histopathological studies, conduction and repolarization disorders in the electrocardiogram, presence of ventricular tachycardia with complete left ventricular bundle branch block morphology in the electrocardiogram or Holter monitoring and family history, especially in first degree relatives with confirmatory genetic test. (1).
Fig. 1. Episode of ventricular tachycardia

Fig. 2. Pathological anatomy of the native heart. Adipose substitution, disorganization, and myocyte fibrosis.
Electrical storm is defined by three or more episodes of sustained ventricular arrhythmia, antitachycardia therapy or ICD shock in the course of 24 h, which predisposes to decompensated heart failure and increased mortality. An initial management with preferably non-selective betablockers is suggested, combined with amiodarone. In case of treatment refractoriness, deep sedation and intubation with mechanical respiratory assistance should be considered to decrease the psychological stress and the pro-arrhythmogenic sympathetic tone. Catheter ablation is associated with reduction of arrhythmia and electrical storm recurrence in patients suitable for this procedure. (2)
According to the 2012 Johns Hopkins registry, including 1000 patients with ACM, only 18 received heart transplantation between 1995 and 2009. They presented at a young age, with left ventricular involvement and 94% survival at one-year post-transplantation. In 13 of these patients, the cause for transplantation was symptomatic heart failure and only in 5, refractory ventricular arrhythmia. (3)
The Nordic registry of cardiac transplantation in patients with ACM, published in 2017, reported 31 transplantations between 1988 and 2014. Compared with a non-transplanted control group with ACM, the only independent risk factor that predicted transplantation was the emergence of the first symptom before 35 years of age. Ninety percent were transplanted for heart failure, compared with 10% for arrhythmia, and with 91% survival at 5 years. (4)
Current heart transplantation guidelines do not provide specific recommendations for ACM due to its low frequency, and because only a small proportion of this population needs to be considered for this procedure. (5) However, the inclusion of patients with refractory ventricular arrhythmias is recommended. (6)
Ethical considerations
Not applicable.
Conflicts of interest
None declared.
(See conflicts of interest forms on the website).
Financing
None.
https://creativecommons.org/licenses/by-nc-sa/4.0/

©Revista Argentina de Cardiología
REFERENCES
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533-41. https://doi.org/10.1161/CIRCULATIONAHA.108.840827
- Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, et al; ESC Scientific Document Group. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43:3997-4126. https://doi.org/10.1093/eurheartj/ehac262
- Tedford RJ, James C, Judge DP, Tichnell C, Murray B, Bhonsale A, et al. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2012;59:289-90. https://doi.org/10.1016/j.jacc.2011.09.051
- Gilljam T, Haugaa KH, Jensen H, Svensson A, Bundgaard H, Hansen J, et al. Heart transplantation in arrhythmogenic right ventricular cardiomyopathy - Experience from the Nordic ARVC Registry. International Journal of Cardiology. 2018;250:201-6. https://doi.org/10.1016/j.ijcard.2017.10.076
- Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, et al; International Society for Heart Lung Transplantation (ISHLT) Infectious Diseases, Pediatric and Heart Failure and Transplantation Councils. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016;35:1-23. https://doi.org/10.1016/j.healun.2015.10.023
- Crespo Leiro MG, Almenar Bonet L, Alonso-Pulpón L, Campreciós M, Cuenca Castillo JJ, de la Fuente Galván L, et al. Conferencia de Consenso de los Grupos Españoles de Trasplante CardiacoRev Esp Cardiol Supl. 2007;7:4B-54B. https://doi.org/10.1016/s1131-3587(07)75240-8