http://dx.doi.org/10.7775/rac.es.v91.i1.20598
ARTÍCULO ORIGINAL
Estudio preliminar
de correlación fenotipo-genotipo en miocardiopatías de pacientes derivados a un
centro de alta complejidad del conurbano bonaerense
A Preliminary Study
of the Phenotype-Genotype Correlation in Cardiomyopathies in Patients Referred
to a Tertiary Healthcare Center in the Suburbs of Buenos Aires
Gisela M. StreitenbergerMTSAC,1,
Graciela R. Reyes1,
Maria P. Velazco1,
Viviana Pasquevich1,
Mariela De Santos1,
Marcos Granillo Fernández1,
Mauricio Potito1,
Pablo Kociubinski2,
Javier MarianiMTSAC,1
1 Servicio
de Cardiología, Hospital de Alta Complejidad en Red El Cruce Néstor Kirchner. Av Calchaquí 5401, Florencio Varela, Provincia de Buenos
Aires, Argentina.
2 Servicio
de Diagnóstico y Tratamiento por Imágenes - Área Cardioresonancia
magnética. Hospital de Alta Complejidad en Red El Cruce Néstor Kirchner. Av Calchaquí 5401, Florencio Varela, Provincia de Buenos
Aires, Argentina.
Dirección para correspondencia: Gisela Streitenberger -
E-mail: gisestreitenberger@yahoo.com.ar
RESUMEN
Introducción: Las miocardiopatías se definen como un trastorno del
miocardio en el que el músculo cardíaco es estructural y funcionalmente
anormal, en ausencia de enfermedad arterial coronaria, hipertensión arterial
(HTA), enfermedad valvular y enfermedad cardíaca congénita. Estas enfermedades
son relativamente frecuentes, y suponen una importante causa de morbimortalidad
a nivel global.
Aunque el estudio genético se recomienda para el
cribado familiar, la falta de datos robustos sobre asociaciones
genotipo-fenotipo específicas ha reducido su impacto en el manejo clínico.
Objetivos: El objetivo de este estudio es analizar la frecuencia
de mutaciones en una población de pacientes con miocardiopatía derivados a un
centro de alta complejidad y el análisis de la correlación genotipo-fenotipo en
las mutaciones identificadas.
Material y métodos: Se
estudiaron en forma prospectiva 102 pacientes con sospecha de miocardiopatía
hipertrófica (MCH) familiar, de los cuales 70 constituían casos índices, de una
cohorte ambispectiva de pacientes con miocardiopatías
controladas en un hospital público de alta complejidad de tercer nivel de
atención de la provincia de Buenos Aires, desde enero 2012 al 30 agosto 2022.
Resultados: De 102 pacientes 83 fueron considerados afectados. De eelos, 31 eran MCH y 52 fenocopias, sin diferencia en el
pronóstico. Se realizó estudio genético en 77 pacientes, de los cuales 57
presentaron mutaciones reconocibles, en el 80% de los casos coincidentes con un
Score de Mayo ≥3. Se detectaron 28 variantes de significado incierto.
Conclusiones: Se comprobó que realizar estudio molecular guiado por
el Score de Mayo permitió obtener un alto grado de probabilidad de detectar
mutaciones. Se evidenció la importancia del estudio molecular debido a la
existencia de solapamiento fenotípico y genotípico de las miocardiopatías. El
conocimiento de la variante genética causal actualmente no afecta el manejo
clínico de la mayoría de los pacientes con MCH, pero es de ayuda ante un pequeño
grupo de genes que tienen opciones de tratamiento.
Palabras
clave: Cardiomiopatías,
Cardiomiopatía Hipertrófica, Sarcómeros, Estudio de
Asociación Genética, Pruebas Genética
ABSTRACT
Background: Cardiomyopathies are defined as a disorder of the
myocardium in which the heart muscle is structurally and functionally abnormal,
in the absence of coronary artery disease, hypertension (HT), valvular heart disease and congenital heart disease. These
diseases are relatively common and a major cause of morbidity and mortality
worldwide.
Although genetic testing is recommended for family
screening, lack of solid data on specific genotype-phenotype associations has
reduced its impact on clinical management.
Objectives: This study aims to analyze the frequency of mutations
in a population of patients with cardiomyopathy referred to a tertiary
healthcare center and to analyze the genotype-phenotype correlation of the
identified mutations.
Methods: We prospectively included 102 patients with suspected
familial hypertrophic cardiomyopathy (HCM), 70 of which were index cases, from
an ambispective cohort of patients with
cardiomyopathies treated in a tertiary healthcare public hospital in the
province of Buenos Aires, from January 2012 to August 30, 2022.
Results: Of 102 patients, 83 were considered affected. Of
these, 31 were HCM and 52 were phenocopies, with no
difference in prognosis. A genetic study was carried out in 77 patients, of
whom 57 presented recognizable mutations, in 80% of the cases coinciding with a
Mayo Score ≥3. Twenty-eight variants of uncertain significance were
detected.
Conclusions: It was confirmed that molecular testing guided by the
Mayo Score provided high probability of detecting mutations. Molecular testing
proved to be important due to the phenotypic and genotypic overlap in
cardiomyopathies. Understanding the causative genetic variant, nowadays, does
not affect the clinical management of most HCM patients, but is helpful in a
small group of genes with treatment options.
Key
words: Cardiomyopathies,
Cardiomyopathy Hypertrophic/genetics, Sarcomeres, Genetic Association Studies,
Genetic Testing
Recibido: 02/12/2022
Aceptado: 30/01/2023
INTRODUCCIÓN
Las miocardiopatías se definen como un trastorno del
miocardio en el que el músculo cardíaco es estructural y funcionalmente
anormal, en ausencia de enfermedad arterial coronaria, hipertensión arterial
(HTA), enfermedad valvular y enfermedad cardíaca congénita. Estas enfermedades
son relativamente frecuentes, y suponen una importante causa de morbimortalidad
a nivel global. (1)
Existen diferentes clasificaciones que pretenden
ayudar a distinguir algunas miocardiopatías de otras, aunque muchas veces
resultan más un factor de confusión que una ayuda en la clasificación. (2,3)
La miocardiopatía hipertrófica (MCH) es una enfermedad
primaria del miocardio producida por mutaciones de genes que codifican
proteínas del sarcómero, con una prevalencia estimada
de hasta 1/200-500 personas, a menudo hereditaria, con una expresión genética y
fenotípica compleja y una historia natural, que afecta a ambos sexos. (4-7)
Se han descrito miles de mutaciones en más de 50 genes
en asociación con la MCH, aunque la frecuencia de mutaciones identificadas es
variable en diferentes estudios y los datos disponibles en nuestro medio son
escasos. (4-7)
Los mecanismos por los cuales las variantes del sarcómero dan como resultado el fenotipo clínico no se han
dilucidado aún. Los genes del sarcómero desencadenan
cambios en el miocardio, que conducen a hipertrofia y fibrosis, un ventrículo
pequeño y rígido con rendimiento sistólico y diastólico deteriorado a pesar de
una fracción de eyección del ventrículo izquierdo (FEVI) conservada. Diversas
características, como arterias coronarias intramurales
anormales responsables de la isquemia y las anomalías de la válvula mitral,
parecen no tener una asociación directa con las variantes del sarcómero. (4-7)
Se cree que los pacientes que carecen de una variante
patogénica tienen MCH no mendeliana y probablemente tienen un mejor pronóstico
que los pacientes con mutaciones patogénicas sarcoméricas.
Identificar la base genética de la MCH crea oportunidades para comprender cómo
se desarrolla la enfermedad y cómo interrumpir su progresión. (5)
Aunque el estudio genético se recomienda para el
cribado familiar, la falta de datos robustos sobre asociaciones
genotipo-fenotipo específicas ha reducido su impacto en el manejo clínico. (5)
El objetivo de este estudio es analizar la frecuencia
de mutaciones en una población de pacientes con miocardiopatía derivados a un
centro de alta complejidad y el análisis de la correlación genotipo-fenotipo en
las mutaciones identificadas.
MATERIAL
Y MÉTODOS
Sujetos
del estudio
Se estudiaron en forma prospectiva 102 pacientes con
sospecha MCH familiar, de los cuales 70 constituían casos índice, de una
cohorte ambispectiva de pacientes con miocardiopatías
controladas en un hospital público de alta complejidad de tercer nivel de
atención de la provincia de Buenos Aires desde enero de 2012 al 30 agosto de
2022.
El diagnóstico de MCH se realizó de acuerdo con los
criterios de la OMS y del grupo de trabajo de enfermedad miocárdica y
pericárdica de la Sociedad Europea de Cardiología. (1)
Los “afectados” de MCH eran aquellos que presentaban 1
criterio mayor electrocardiográfico (ECG) o en la ecocardiografía transtorácica (ETT), o 2 criterios menores ETT más 1 menor
ECG, o 2 criterios menores ECG más 1 menor ETT. (7)
A los pacientes con diagnóstico de miocardiopatía se
les realizó historia clínica y familiar incluyendo 3 generaciones, exploración
física, genograma, ECG, ETT, ECG Holter (en
afectados), ergometría o test cardiopulmonar de esfuerzo (en afectados),
hemograma y bioquímica completa con NT-proBNP y Troponina (en afectados).
Parámetros
ecocardiográficos
Los estudios se realizaron con un Ecocardiógrafo
Epiq 7 CVx 3D (Philips
Medical Systems) utilizando un transductor S5-1. Se
obtuvieron mediciones de la FEVI y función diastólica de acuerdo con las recomendaciones
de la Sociedad Americana de Ecocardiografía. (8,9)
Una FEVI <52% en hombres y <54% en mujeres fue
considerada deprimida.
El análisis de Speckle-tracking
se realizó de acuerdo con las recomendaciones vigentes del consenso
EACI/ASE. (10) Los bucles de cine a partir de tres
vistas apicales del VI estándar (cuatro, dos y tres cámaras) se registraron
utilizando imágenes con armónicas en escala de grises, con la mayor velocidad
de cuadros posible (55-90 cuadros/seg). El análisis
de los archivos grabados se realizó off line por un ecocardiografista
experimentado no cegado al diagnóstico del paciente.
El strain longitudinal
global (SLG) por 2D se evaluó en 16 segmentos promediados del VI (Software para
el post-procesado del strain: TOMTEC. Dynamic Heart Model).
El operador ajustó manualmente la región de interés en segmentos que no se pudo
rastrear correctamente. Valor normal de SLG global Philips: -21± 2%.
Cardioresonancia magnética (CRM)
Se empleó un equipo Philips Medical Systems Achieva Serie X de 3T. Se
tomaron imágenes anatómicas del corazón con secuencias Sangre Negra y Sangre
Blanca. Se realizó estudio funcional con imágenes de cine gatilladas. Se
realizaron adquisiciones con secuencias de realce en T2 y en T1, supresión
grasa, secuencia con TE variable y Tagging. Con la
inyección de gadolinio (dosis total: 0,2 mmol/kg) se
realizaron secuencias de primer paso (0,1 mmol/kg) e
imágenes tardías (Realce Tardío), que luego fueron post-procesadas y evaluadas
con el programa Extended MR Space 2.6.3.3.
Análisis
mutacional
Se utilizó la puntuación de predictor de genotipos de
HCM de Mayo (Score de Mayo), para predecir el rendimiento diagnóstico de las
pruebas genéticas y guiar el uso del método NGS (secuenciación de nueva
generación). (11,12)
Se realizó test molecular a aquellos con Score de Mayo
≥3 (rango de -1 a 5) o a los familiares de pacientes con mutaciones positivas.
Se invitó a familiares de primer grado a realizar un estudio clínico, ECG y
ETT, para identificar a los afectados, a los que se les ofreció realizar
estudio genético.
El componente técnico de la secuenciación
confirmatoria lo realizó Invitae Corporation a partir de muestras de saliva recogidas
mediante un hisopado bucal. La clasificación de variantes identificadas se
realizó con pautas del American College of Medical
Genetics and Genomics. (13)
Todos los pacientes firmaron un consentimiento
informado y el estudio fue aprobado por el comité de ética de la institución.
Estudio
de la correlación genotipo-fenotipo
Para la descripción de las características fenotípicas
se utilizó la clasificación de Maron con los cuatro
fenotipos clásicos, según la ubicación y el grado de hipertrofia. (14)
Para correlacionar fenotipo (F) con genotipo (G) se
utilizó la clasificación de Lever que permite evaluar
la probabilidad pretest según subtipo anatómico. (15)
Se definió MCH obstructiva cuando presentaba un
gradiente de presión intraventricular significativo (≥30
mmHg) en reposo, obstructiva latente cuando el
gradiente se evidenciaba después de maniobras de provocación (Valsalva/ de pie/ejercicio) y no obstructivas a aquellas
con gradiente < 30 mmHg.
Definiciones en ANEXO.
Eventos
cardiovasculares
Se definió como eventos cardiovasculares (ECV) la
presencia o ausencia de los siguientes:
• Necesidad de colocación de cardiodefibrilador
implantable (CDI) o marcapasos
• Muerte súbita
• Internación por causa cardiovascular
• Miectomía septal o ablación septal con
alcohol
El seguimiento se realizó durante 3 años posterior al
diagnóstico mediante controles clínicos por consultorios externos o llamados
telefónicos.
Análisis
estadístico
Se utilizó el programa estadístico Epi
Info para PC versión 7.2.4.0 y Statistix
7.
Las variables cualitativas se describieron utilizando
números y porcentajes. Las variables cuantitativas se describieron utilizando
media y desvío estándar o mediana y rango intercuartilo
(RIC), dependiendo de si la distribución era normal o no respectivamente.
Para las comparaciones entre grupos se utilizó el test
de Student para variables continuas con distribución
normal, y pruebas no paramétricas (U de Mann-Whitney) para variables continuas
con distribución no normal; y test de Chi cuadrado (χ²) o test exacto de
Fisher en el caso de variables categóricas. Se consideró estadísticamente
significativo un valor de p < 0,05.
RESULTADOS
Se evaluaron 102 pacientes. El diagrama del flujo
diagnóstico se presenta en la Figura 1.
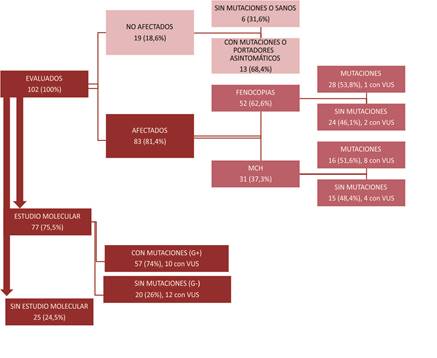
MCH: miocardiopatía hipertrófica VUS:
variantes de significado incierto
Fig. 1. Diagrama de flujo diagnóstico
La presencia de fenotipo compatible con MCH se
determinó en base a los criterios eléctricos y ecocardiográficos.
Se clasificó a los pacientes en dos grupos: “afectados” (n = 83, 81,4%, IC 95%
72,4-88,4%) y “no afectados” (19, 18,6%, IC95% 11,6- 27,5%) (Tabla
1A).
Tabla 1. Características de la población estudiada. A. Parámetros clínicos y electrocardiografía
|
PARÁMETROS
CLÍNICOS |
Pacientes (N=102) |
Afectados (n=83) |
No
afectados (n=19) |
|
|
Edad actual (años) |
45 ± 16 |
47,6 ± 16 |
33,7 ± 10 |
|
|
Edad Diagnóstico (años) |
41,9 ± 16 |
28,8 ± 14 |
0,001 |
|
|
Sintomáticos |
83 (97,6%) |
81 (97,6%) |
2 (10,5%) |
|
|
Sexo femenino |
56 (54,9%) |
40 (48,2%) |
16 (84,2%) |
0,003 |
|
Antecedentes familiares |
71 (69,6%) |
52 (62,6%) |
19 (100%) |
≤0,001 |
|
Peso (kg) |
70 ± 18 |
70 ± 18 |
69 ± 18 |
0,9 |
|
Hipertensión arterial |
24 (23,5%) |
23 (27,7%) |
1 (5,3%) |
0,02 |
|
TAS (mmHg) |
107 ± 16,8 |
105 ± 16 |
117 ± 14 |
0,005 |
|
Obesidad |
22 (21,5%) |
18 (21,7%) |
4 (21%) |
0,6 |
|
FC (latidos/minuto) |
71,6 ± 15 |
71,3 ± 16 |
73,2 ± 8,3 |
0,62 |
|
Diabetes |
8 (7,8%) |
7 (8,4%) |
1 (5,2%) |
0,53 |
|
Dislipidemia |
10 (9,8%) |
10 (12%) |
0 |
0,11 |
|
Disnea CF ≥II NYHA |
80 (78,4%) |
80 (96,4%) |
0 |
<0,001 |
|
Ángor |
38 (37,2%) |
37 (44,5%) |
1 (5,3%) |
<0,001 |
|
Síncope |
50 (49%) |
48 (57,8%) |
2 (10,5%) |
<0,001 |
|
Enfermedad coronaria |
4 (3,9%) |
4 (4,8%) |
0 |
0,43 |
|
Valor BNP (pg./mL) |
|
986 (122-3237) |
|
|
|
Troponina I |
|
12,5 (4-59,5) |
|
|
|
Electrocardiograma |
|
|
|
|
|
Anormal |
88 (86,3%) |
82 (98,8%) |
6 (31,6%) |
|
|
T negativas |
34
(33,3%) |
33
(39,7%) |
1
(5,3%) |
<0,001 |
|
Signos
HVI |
54 (52,9%) |
51 (61,4%) |
3 (15,8%) |
<0,001 |
|
BCRI |
12
(11,7%) |
12
(14,4%) |
0 |
0,07 |
|
BCRD |
4 (3,9%) |
4 (4,8%) |
0 |
0,43 |
|
HBAI |
23(22,5%) |
23
(27,7%) |
0 |
<0,001 |
|
Patron QS |
52 (50,9%) |
51 (61,4%) |
1 (5,3%) |
<0,001 |
|
Microvoltaje |
25
(24,5%) |
25
(30,1%) |
0 |
0,01 |
|
Ritmo sinusal |
92 (90,2%) |
73 (87,9%) |
19 (100%) |
0,24 |
|
FA parox. o permanente |
18
(17,6%) |
18
(21,7%) |
0 |
0,01 |
|
Taquicardia
ventricular |
12 (11,7%) |
12 (14,4%) |
0 |
0,07 |
|
Ecocardiografia |
|
|
|
|
|
FEVI normal |
78 (76,5%) |
59 (71%) |
19 (100%) |
<0,001 |
|
FEVI (%) |
62
(55-66) |
60
(52-67) |
66
(61-70) |
0,03 |
|
Hipertrofia
VI |
61 (59,8%) |
61 (73,5%) |
0 |
<0,001 |
|
Dilatación
AI |
79 (77,4%) |
76 (91,5%) |
3 (15,8%) |
<0,001 |
|
Disfunción diastólica |
82 (80,4%) |
80 (96,4%) |
2 (10,5%) |
<0,001 |
|
Relación
E/e´ |
11,6 ± 4,6 |
12,7 ± 4,2 |
6,6 ± 1,7 |
<0,001 |
|
SLG promedio (%) |
17 (12-20) |
16 (10-19) |
22 (20-22) |
<0,001 |
AI: aurícula izquierda; BCRD:
bloqueo completo de rama derecha; BCRI: bloqueo completo de rama izquierda;
BNP: péptido natriurético B; CF: clase funcional; FA:
fibrilación auricular; HBAI: hemibloqueo anterior
izquierdo; HVI: hipertrofia ventricular izquierda; FEVI: fracción de eyección
del ventrículo izquierdo; FSVI: Función sistólica ventricular izquierda; parox: paroxística;
SLG: strain longitudinal global; TAS: tensión
arterial sistólicaLas variables cualitativas se
presentan como n (%), y las cuantitativas como media ± desviación estándar o
mediana y rango intercuartilo
La edad media de diagnóstico de los síntomas fue de 39
± 16,7 años, con inicio más temprano en las mujeres (34,7± 15 años; p< 0,001),
aquellos con antecedentes familiares (36,7± 16 años; p< 0,001) y variantes
patogénicas en TNNT2.
Entre los afectados la presencia de síntomas fue más
frecuente en los hombres (n = 43) que en las mujeres (n = 40) y el síntoma más
frecuente fue la disnea.
Se realizó ECG y ETT al 100% de los pacientes, y CRM a
64 (62,7%). No se realizó CRM por claustrofobia, negación y presencia de
dispositivos cardíacos.
En la Tabla 1B se muestran las
características de los 83 “afectados”. El 71% (n=59) tenía FEVI conservada,
frente al 100% de los no afectados; p < 0,001. El 37,4% (n = 31) eran MCH y
el 62,6% (n = 52) fenocopias. Aproximadamente la mitad de las MCH eran obstructivas
(51,6%) y la mayoría tenía FEVI conservada (25, el 80%). El SLG promedio de los
pacientes con amiloidosis fue de 14% (9-17)
estadísticamente diferente (p = 0,01) al de los pacientes con MCH, y el índice
FEVI/SLG con un valor de corte ≥4,3 ± 1,6 permitió diferenciar amiloidosis de MCH (p <0,001), como en estudios
anteriores. (16)
Tabla 1. Características de la población estudiada. B. Características ecocardiográficas y de cardioresonancia
de la población “afectada”
|
Ecocardiográficos |
Afectados n=83 |
MCH n=31 |
Fenocopia n=52 |
|
|
|
FEVI
NORMAL |
59 (71%) |
25 (80,6%) |
34 (65,4%) |
0,10 |
|
|
FEVI
(%) |
60 (52-67) |
64 (55-69) |
58 (44-66) |
0,09 |
|
|
Espesor SIV (mm) |
13 (9-17) |
18 (15-28) |
12 (9,5-15) |
<0,001 |
|
|
IMVI
(gr/m2) |
109 (78-141) |
138 (119-185) |
109 (86- 133) |
0,001 |
|
|
OTSVI |
16 (19,3%) |
16 (51,6%) |
0 |
<0,001 |
|
|
Dilatación AI |
76
(91,9%) |
30
(96,7%) |
46
(88,4%) |
0,18 |
|
|
I.
Vol AI (ml/m2) |
41
(32-56) |
51,5
(43-82,5) |
40
(36-55) |
0,003 |
|
|
80 (96,4%) |
31 (100%) |
49 (94,2%) |
0,24 |
||
|
1-
Relajación prolongada |
8 (9,6%) |
0 |
8 (15,4%) |
0,1 |
|
|
2-
Pseudonormal |
57
(67,5%) |
24
(77,4%) |
32
(61,5%) |
0,1 |
|
|
3-
Restrictivo |
11
(13,2%) |
3 (9,7%) |
8 (15,4%) |
0,1 |
|
|
4-
Monofásico |
8 (9,6%) |
4 (12,9%) |
3 (5,7%) |
0,1 |
|
|
E/e´ |
12,7 ±
4,2 |
13,4 ±
3,9 |
12,3 ±
4,4 |
0,27 |
|
|
Membrana subaórtica |
2 (2,4%) |
2(6,4%) |
0 |
0,13 |
|
|
VAo bicúspide |
2 (2,4%) |
2(6,4%) |
0 |
0,13 |
|
|
Insuficiencia aortica |
16
(19,3%) |
10
(32,2%) |
6 (11,5%) |
0,02 |
|
|
Insuficiencia mitral |
63 (76%) |
26
(83,8%) |
37
(71,1%) |
0,19 |
|
|
Insuficiencia tricuspídea
|
68 (82%) |
28
(90,3%) |
40 (77%) |
0,10 |
|
|
PSAP |
27 (0-39) |
33
(25-43) |
30 (0-40) |
0,15 |
|
|
SLG
promedio (%) |
16
(10-19) |
17
(13-19) |
15 (9-17) |
<0,001 |
|
|
FEVI/SLG |
3,7 (3,3-
4,7) |
3,6 (3,2-
4,2) |
3,9 (3,4-
5,3) |
0,27 |
|
|
CARDIORESONANCIA |
63
(75,9%) |
25
(80,6%) |
38 (73%) |
0,43 |
|
|
Masa
ventricular indexada (gr/m2) |
80 (62-
96) |
85
(62-122) |
73
(62-90) |
0,24 |
|
|
FEVI
(%) |
61
(44-71) |
70
(59-73) |
60
(40-67) |
<0,001 |
|
|
FEVD
(%) |
72
(61-78) |
71 (64-81) |
72
(60-77) |
0,34 |
|
|
RTG |
44
(72,1%) |
18
(85,7%) |
26
(65%) |
0,07 |
|
AI: aurícula izquierda; BCRD:
bloqueo completo de rama derecha; BCRI: bloqueo completo de rama izquierda;
BNP: péptido natriurético B; CF: clase funcional; FA:
fibrilación auricular; HBAI: hemibloqueo anterior
izquierdo; HVI: hipertrofia ventricular izquierda; FEVI: fracción de eyección
del ventrículo izquierdo; FSVI: Función sistólica ventricular izquierda; parox: paroxística;
SLG: strain longitudinal global; TAS: tensión
arterial sistólicaLas variables cualitativas se
presentan como n (%), y las cuantitativas como media ± desviación estándar o
mediana y rango intercuartilo
Mutaciones
identificadas
Se realizó estudio molecular a 77 pacientes (75,5%),
de los cuales 57 (el 75%) presentaban mutaciones (G+) y 20 (26%) no (G-). De
los 57 pacientes con G+, 46 (80,7%) tenían un Score de Mayo ≥3; p
<0,001 vs. los pacientes con G-. Se detectaron 22 (28,5%) variantes de
significado incierto (VUS, por sus siglas en inglés) (Figuras 2
y 3). Dos pacientes presentaban 2 variantes patogénicas en heterocigosis y 10 presentaban VUS además de la mutación
patogénica.
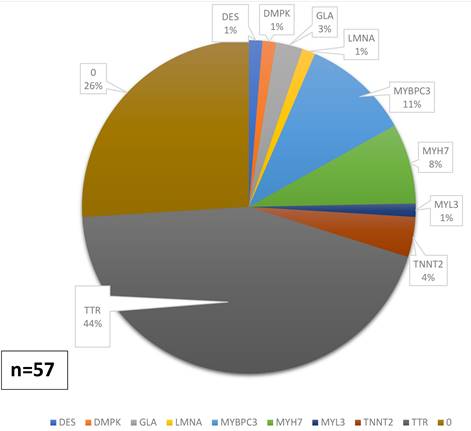
Fig. 2. Variantes genéticas identificadas
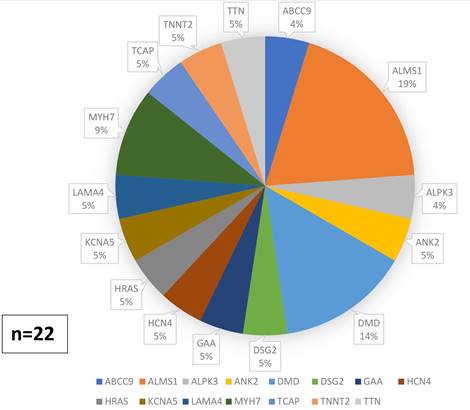
Fig. 3. Variantes genéticas de significado incierto (VUS) identificadas
Entre los 19 no afectados se pudo descartar la
enfermedad en 6 (31,6%), mientras que 13 (68,4%) fueron portadores
asintomáticos. La penetrancia de la enfermedad
(“afectados con mutaciones”) fue del 77,2% (44 de los 57 con G+); de los 44, 16
(36,4%) eran MCH.
Correlación
Fenotipo- Genotipo
La forma de presentación más frecuente fue el tipo 1 y
3 de Maron (afección septal
y anterolateral), con más G+ detectados en la tipo 1
en la MCH (9, el 75%) y en la tipo 3 en las fenocopias (13, el 68,4%),
p<0,001.
La clasificación de Lever 2
y 4 (septum reverso y neutro, respectivamente) resultó de gran utilidad a la
hora de valorar la probabilidad de presentar un G+ basado en el fenotipo (F)
anatómico expresado por el paciente (Tabla 2 y Figura 4).
Tabla 2. Correlación fenotipo-genotipo de los pacientes con estudio molecular
|
Fenotipos |
Pacientes n=77 |
|
Mutaciones (G+) n=57 |
|
Sin
mutaciones (G-)
n=20 |
|
p |
|
Fenocopias |
42
(54,5%) |
|
33
(57,8%) |
|
9
(45%) |
|
0,46 |
|
MCH |
21 (27,3%) |
|
16 (28%) |
|
5 (25%) |
|
0,52 |
|
MARON 1 |
17
(22%) |
|
13
(23%) |
|
4
(20%) |
|
0,53 |
|
MARON 2 |
6 (7,8%) |
|
5 (8,7%) |
|
1 (5%) |
|
0,88 |
|
MARON 3 |
21
(27,3%) |
|
14
(24,6%) |
|
7 (35%) |
|
0,26 |
|
MARON 4 |
6 (7,8%) |
|
5 (8,7%) |
|
1 (5%) |
|
0,88 |
|
LEVER 1 |
1
(1,3%) |
|
0 |
|
1 (5%) |
|
0,39 |
|
LEVER 2 |
18 (23,4%) |
|
14 (24,5%) |
|
4 (20%) |
|
0,46 |
|
LEVER 3 |
1
(1,3%) |
|
1
(1,75%) |
|
0 |
|
0,39 |
|
LEVER 4 |
25 (32,4%) |
|
17 (29,8%) |
|
8 (40%) |
|
0,39 |
|
Edad diagnóstica |
38
(29-48) |
|
36
(28-44) |
|
45,5 (37,5-51,5) |
|
0,05 |
|
Sexo femenino |
46 (59,7%) |
|
36 (63,1%) |
|
10 (50%) |
|
0,44 |
|
HTA |
15
(19,5%) |
|
8
(14%) |
|
7 (35%) |
|
0,04 |
|
Eventos CV |
33 (42,8%) |
|
22 (38,6%) |
|
11 (55%) |
|
0,31 |
|
Trasplante cardíaco |
1 (4%) |
|
1
(2,2%) |
|
0 |
|
0,44 |
|
Taquicardia ventricular |
14 (13,8%) |
|
5 (8,7%) |
|
3 (15%) |
|
0,34 |
|
CDI |
16
(20,8%) |
|
11
(19,3 %) |
|
5 (25%) |
|
0,40 |
|
Marcapaso |
12 (15,6%) |
|
8 (14%) |
|
4 (20%) |
|
0,37 |
|
Miectomía |
6
(7,8 %) |
|
4
(7%) |
|
2 (10%) |
|
0,49 |
|
Internación CV |
26 (33,7%) |
|
16 (28%) |
|
10 (50%) |
|
0,06 |
|
Muerte CV |
11
(14,3%) |
|
8
(14%) |
|
3 (15%) |
|
0,58 |
CV: cardiovascular; CDI: cardiodefibrilador implantable;
HTA: hipertensión arterial; MCH: miocardiopatía hipertrófica
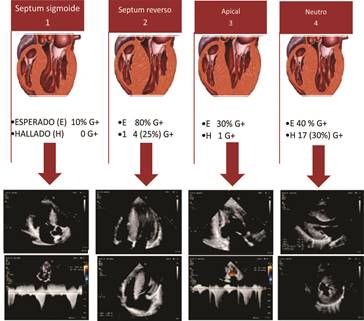
Imagen de ecocardiografía Doppler color de fuente propia. Figura
modificado de: Lever HM, Karam
RF, Currie PJ, Healy BP. Hypertrophic cardiomyopathy in the elderly. Distinctions
from the Young based on cardiac
shape. Circulation 1989;
79(3):580-9.
Fig. 4. Patrones de hipertrofia según la clasificación de Lever
et al. Probabilidad pretest de estudio genético
positivo según subtipo anatómico
No hubo diferencias significativas en ECV, tratamiento
médico o intervenciones realizadas entre los pacientes con G+ o G- (Figura 5).

Fig. 5. Curva de Kaplan-Meier de sobrevida libre de
eventos. Los p con G+ tienen un HR de mortalidad de 1,67 respecto de los G-, p
= 0,14
La mediana de tiempo desde el inicio de la miocardiopatía
hasta un ECV fue de 2,4 años. No se produjeron mas
ECV en los pacientes con fenocopias (29, el 55,7%) que en las MCH (17, el
54,8%), p = 0,93. Se produjeron más muertes cardiovasculares en los hombres
(10, 21,7%) que las mujeres (3, 5,3%); p = 0,01.
DISCUSIÓN
Las miocardiopatías son un grupo heterogéneo de
enfermedades del miocardio asociadas con disfunción mecánica y/o eléctrica que
generalmente presentan una hipertrofia o dilatación ventricular inapropiada y
se deben a una variedad de causas, frecuentemente genéticas. (1)
Para que la genética sea una herramienta útil en la
toma de decisiones clínicas es necesario disponer de información detallada
sobre las características clínicas y morfológicas de los portadores de las
diferentes mutaciones, como la que proporciona este estudio.
El Score de Mayo permitió elegir mejor a los probandos y que el estudio molecular sea costo-efectivo,
con la limitación financiera evidente.
La remodelación hipertrófica también ocurre en
trastornos que clínicamente imitan a la MCH, incluida la enfermedad de Fabry (mutaciones en GLA) y la amiloidosis
por transtiretina (TTR) entre otros. Se han informado
más de 1500 mutaciones en al menos 8 genes de proteínas del sarcómero
en la MCH, aunque la mayoría (80%) de las mutaciones alteran la cadena pesada
de B-miosina (MYH7) o el gen de la proteína C ligada a miosina (MYBPC3). El
diverso origen molecular combinado con la variabilidad genómica de fondo y las
diferencias en el estilo de vida entre los pacientes ha dificultado la
comprensión definitiva de las relaciones genotípicas y fenotípicas. (4)
Estudios previos sugieren que las mutaciones en MYH7
causan entre un 15-30% de los casos de MCH. (17,18)
En nuestros pacientes, las mutaciones en este gen son menos frecuentes y
aparecen en un 7,5% de las familias estudiadas, esta diferencia puede deberse
al grado de selección de la población estudiada.
Aunque no encontramos diferencias significativas en la
edad en el momento del diagnóstico, la edad media de los pacientes con G+ fue
de 37,4 ± 15 años, frente a 42,4 ± 18 años en los G-, similar a otras series. (19,20)
Un dato interesante de nuestro estudio fue la mayor
frecuencia de mutaciones identificadas en mujeres (36, 63%; p=0,06), sin
significancia estadística; pero como el patrón de herencia habitual en la MCH
es autosómico dominante cabría esperar que un 50% de los pacientes fueran mujeres.
Sin embargo, en prácticamente todas las series descritas, la proporción de
mujeres ronda entre el 30 y el 40%, y suelen ser mayores en el momento del
diagnóstico. En nuestro estudio ocurrió lo contrario (edad de diagnóstico de
las mujeres: 37 ± 15 años, frente a 46,5 ± 15 años en los hombres; p<0,01). (17-20)
Las mujeres tuvieron una mayor prevalencia del
fenotipo obstructivo, síntomas más graves con requerimiento de terapia de
reducción septal e incluso una paciente se trasplantó.
Sin embargo, hubo mayor mortalidad en hombres que en mujeres.
La identificación de mutaciones en diferentes familias
permite hacer una valoración más precisa de la correlación genotipo-fenotipo y
la interpretación adecuada del papel patogénico de cada mutación. Varios
hallazgos de nuestro estudio inciden en la importancia de realizar un estudio
familiar completo. Mientras que en algunas mutaciones, como la TNNT2
c.812A>T (p.Asn271Ile), el fenotipo se reproduce de forma similar en la
mayor parte de los portadores, en otras, como la MYBPC3 c.1808_1821del
(p.Ile603Thrfs*6), llama la atención la gran diferencia entre el fenotipo de
los casos índice (hipertrofia severa en pacientes jóvenes) y los familiares
portadores con hipertrofia ligera, a pesar de tener edades similares o mayores.
En estos casos debe considerarse la posibilidad de que haya factores genéticos
o ambientales adicionales que expliquen la gran diferencia de expresión. En
varios trabajos se ha demostrado que los pacientes con MCH pueden presentar más
de una mutación y que la presencia de dobles mutaciones se asocia con una
expresión más severa de la enfermedad como es el caso de dos pacientes de
nuestro estudio. (6, 17-20)
En la práctica clínica es frecuente la coincidencia de
la MCH con hipertensión arterial (8 pacientes con MCH en nuestro estudio, el
25,8%) debido a la alta prevalencia de ambas enfermedades. Esta situación
hemodinámica modifica necesariamente el fenotipo de la MCH como también el
ejercicio (corazón del atleta) y otras comorbilidades (diabetes mellitus,
obesidad e insuficiencia renal crónica), está claro que a menudo es imposible
distinguir la causa real o el principal modificador de la hipertrofia del VI.
Actualmente, se cree que la amiloidosis
TTR de tipo natural o wild-type (wt), intensamente estudiada y subdiagnosticada,
tiene una prevalencia relativamente alta. En nuestro estudio se detectaron 46
pacientes con amiloidosis cardíaca (45%) de los
cuales 60,7% fueron amiloidosis TTRv
(variante), 26% TTRwt, y el resto otras amiloidosis. Creemos que la amiloidosis
fue el factor que contribuyó a la mayor mortalidad en el grupo “fenocopias” con
respecto al grupo MCH.
Limitaciones
Estudio realizado en un solo centro de derivación de
tercer nivel de atención con individuos probablemente más graves y
sintomáticos.
Podría haber alguna mutación adicional que no hubiera
sido identificada debido al uso de paneles predeterminados de genes.
La recogida de muestras no es posible en sujetos
fallecidos ni en los que han declinado participar en el estudio o no han sido
avisados por el caso índice.
CONCLUSIONES
Se comprobó que realizar estudio molecular guiado por
el Score de Mayo permitió obtener un alto grado de probabilidad de detectar
mutaciones. Se evidenció la importancia del estudio molecular debido a la existencia
de solapamiento fenotípico y genotípico de las miocardiopatías.
El conocimiento de la variante genética causal
actualmente no afecta el manejo clínico de la mayoría de los pacientes con MCH,
pero es de ayuda en un pequeño grupo de genes, como GAA, GLA, LAMP2, PRKAG2 y
TTR, que están indiscutiblemente asociados con enfermedades que imitan la MCH
y tienen perfiles clínicos, patrones de herencia y opciones de tratamiento
distintivos, por lo cual, en estos casos, el estudio molecular representa un
paso significativo hacia enfoques personalizados.
Declaración
de conflicto de intereses
Los autores declaran que no tienen conflicto de
intereses
(Véanse formularios de conflicto de intereses de los
autores en la web/Material suplementario).
1. Elliott P, Andersson B, Arbusteini,
Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a
position statement from the european society of
cardiology working group on myocardial and pericardial diseases, Eur Heart J 2008;29:270–6 https://doi.org/10.1093/eurheartj/ehm342
2. Thiene G, Corrado
D, Basso C. Revisiting definition and classification of cardiomyopathies in the
era of molecular medicine. Eur Heart J 2008;29:144–6. https://doi.org/10.1093/eurheartj/ehm585
3. Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Diagnosis and Evaluation of Hypertrophic
Cardiomyopathy. J Am Coll Cardiol.
2022;79:372–89. https://doi.org/10.1016/j.
jacc.2021.12.002
.
4. Ommen SR, Mital
S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020
AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic
cardiomyopathy: a report of the American College of Cardiology/American Heart
Association Joint Committee on Clinical Practice Guidelines. Circulation 2020;142:e558–e631. https://doi.org/10.1161/CIR.0000000000000937
5. Mazzarotto F, Olivotto
I, Boschi B, Girolami F, Poggesi C, Barton PJR, et al. Contemporary Insights Into
the Genetics of Hypertrophic Cardiomyopathy: Toward a New Era in Clinical
Testing? J Am Heart Assoc 2020;0:e015473.
https://doi.org/10.1161/JAHA.119.015473
6. Maron BJ, Maron
MS, Semsarian C. Genetics of Hypertrophic
Cardiomyopathy After 20 Years: Clinical Perspectives. J Am Coll
Cardiol 2012;60:705-15. https://doi.org/10.1016/j.jacc.2012.02.068
7. McKenna WJ, Spirito P, Desnos
M, Dubourg O, Komajda M.
Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for
new diagnostic criteria in adult members of affected families. Heart 1997;77:130-2. https://doi.org/10.1136/hrt.77.2.130
8. Lang RM, Badano LP, Mor-Avi
V, Afilalo J, Armstrong A, Ernande
L, et al. Recommendations for cardiac chamber quantification by
echocardiography in adults: an update from the American Society of
Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2015;28:1-39. https://doi.org/10.1016/j.echo.2014.10.003
9. Nagueh SF, Smiseth
OA, Appleton CP, Byrd BF 3rd, Dokainish H, Edvardsen T, et al. Recommendations for the evaluation of
left ventricular diastolic function by echocardiography: an update from the
American Society of Echocardiography and the European Association of Cardiovascular
Imaging. J Am Soc Echocardiogr
2016;29:277-314. https://doi.org/10.1016/j.echo.2016.01.011
10. Voigt JU, Pedrizzetti G, Lysyansky P, Marwick TH, Houle H,
Baumann R, et al. Definitions for a common standard for 2D speckle tracking
echocardiography: consensus document of the EACVI/ ASE/Industry Task Force to
Standardize Deformation Imaging. J Am Soc Echocardiogr 2015;28:183-93. https://doi.org/10.1016/j.echo.2014.11.003
11. Bonaventura J, Norambuena P, Tomašov P, Jindrová D, Šedivá H, Macek M Jr, et.al. The
utility of the Mayo Score for predicting the yield of genetic testing in
patients with hypertrophic cardiomyopathy. Arch Med Sci. 2019;15:641-9.
https://doi.org/10.5114/aoms.2018.78767
12. Bos J.M., Will M.L., Gersh
B.J., Kruisselbrink T.M., Ommen
S.R., Ackerman M.J. Characterization of a Phenotype-Based Genetic Test
Prediction Score for Unrelated Patients With Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2014;89:727–37. https://doi.org/10.1016/j.mayocp.2014.01.025
13. Bennett RL, Steinhaus KA, Uhrich SB, O’Sullivan CK, Resta
RG, Lochner-Doyle D, et al. Recommendations for
standardized human pedigree nomenclature. Pedigree Standardization Task Force
of the National Society of Genetic Counselors. Am J Hum Genet. 1995;56:745-52.
14. Maron BJ. Hypertrophic cardiomyopathy. Curr Probl Cardiol
1993;18:639-704. https://doi.org/10.1016/0146-2806(93)90025-w
15. Lever HM, Karam RF, Currie PJ, Healy BP.
Hypertrophic cardiomyopathy in the elderly. Distinctions from the young based
on cardiac shape. Circulation 1989;79:580-9. https://doi.org/10.1161/01.CIR.79.3.580
16. Saad A, Arbucci
R, Rousse G, Darú V, Merlo
P, Lowenstein J y col. Perfiles ecocardiográficos
del strain 2D permiten diferenciar a la amiloidosis cardíaca de la miocardiopatía hipertrófica con
fracción de eyección conservada. Rev Argent Cardiol 2018;86:20-6. https://doi.org/10.7775/rac.v86.i6.14239
17. Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. ACC/ESC
clinical expert consensus document on hypertrophic cardiomyopathy: a report of
the American College of Cardiology Task Force on Clinical Expert Consensus
Documents and the European Society of Cardiology Committee for Practice
Guidelines (Committee to Develop an Expert Consensus Document on Hypertrophic
Cardiomyopathy). Eur Heart J 2003;24:1965-9.
https://doi.org/10.1016/S0195-668X(03)00479-2
18. Laredo R, Monserrat L, Hermida-Prieto M, Fernández X, Rodríguez I,
Cazón L, et al. Mutaciones en el gen de la cadena pesada de la betamiosina en la miocardiopatía hipertrófica. Rev Esp Cardiol
2006;59:1008-18. https://doi.org/10.1157/13093977
19. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichreau C, et al. Hypertrophic cardiomyopathy:
distribution of disease genes, spectrum of mutations, and implications for a
molecular diagnosis strategy. Circulation 2003;107:2227-32 http://doi.org/10.1161/01.CIR.0000066323.15244.5
20. García-Castro M, Cotoa E, Reguerob JR, Berrazuetac JR, Álvareza V, Alonso B, et al. Mutaciones en genes sarcoméricos en la miocardiopatía hipertrófica. Rev Esp Cardiol.
2009;62:48-5. https://doi.org/10.1016/S0300-8932(09)70020-X65
ANEXO
1
Criterios diagnósticos de MCH. World Health
Organization/International Society and Federation of Cardiology.1997
|
Criterios Menores |
|
|
Ecocardiográficos (ETT) |
|
|
- Septo anterior o pared posterior
≥ 13 mm - Septo posterior o pared libre ≥
15 mm - SAM severo |
- Septo anterior o pared posterior ≥ 12
mm - Septo posterior o pared libre ≥ 14mm - SAM moderado - Velos mitrales redundantes |
|
Electrocardiográficos (ECG) |
|
|
- HVI + alteraciones en la
repolarización - Inversión de onda T en derivaciones
I y aVL, V3-V6 (≥ 3 mm), o
II, III, aVf (≥ 5 mm) - Ondas Q anormal (> 40 ms o> 25% de onda R) en al menos 2 derivaciones de II, III, aVF, V14 o I
aVL o V5-6 |
- BCRIHH o trastorno de la conducción
intraventricular en derivaciones izquierdas. - Alteraciones en la repolarización menores
en derivaciones izquierdas - Ondas S profundas en V2 (> 25 mm) |
|
Clínicos |
Sincope innexplicable, disnea o
dolor precordial |
|
Diagnóstico de Miocardiopatía hipertrofica |
|
|
1 criterio
mayor, ó |
2 criterios menores ETT + 1 criterio menor ECG |
Cuadro modificado de McKenna WJ, Spirito
P, Desnos M, Dubourg O, Komajda M. Experience from clinical genetics in
hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult
members of affected familyes. Heart 1997;77(2):130-132
Criterios
diagnósticos de MCH de ESC.2008
Adultos
Grosor de la pared ≥15 mm en uno o más segmentos
miocárdicos del VI –determinado por cualquier técnica de imagen:
ecocardiografía, imagen por cardioresonancia magnética
(CRM) o tomografía computarizada (TC)– que no puede explicarse únicamente por
condiciones de carga
Niños
Engrosamiento de la pared del VI con un Z score >2
desviaciones estandar de la media esperable.
Familiares
Presencia inexplicable de un aumento del grosor del VI
≥13 mm en uno o más segmentos miocárdicos del VI, medido por cualquier
técnica de imagen (ecocardiografia, CRM o TC).
Tomado de Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the
European Society Of Cardiology Working Group on Myocardial and Pericardial
Diseases. Eur Heart J 2008;29(2):270-6
– “Fenocopias de MCH (Imitaciones)”:
enfermedades cardíacas o sistémicas capaces de producir HVI que no deben
etiquetarse como MCH. El uso de MCH para describir el aumento del grosor de la
pared del VI asociado con trastornos sistémicos o causas secundarias de
hipertrofia del VI (HVI) puede generar confusión. Los trastornos sistémicos
incluyen: síndromes metabólicos y multiorgánicos como
las RASopatías (variantes en varios genes implicados
en la señalización de RAS-MAPK), miopatías mitocondriales, enfermedades de
almacenamiento de glucógeno/lisosoma en niños y miocardiopatía de Fabry, amiloidosis, sarcoidosis, hemocromatosis y Danon
en adultos. En estas enfermedades, aunque la magnitud y distribución del
aumento del grosor de la pared del VI pueden ser similares a las de la MCH
aislada causada por variantes en los genes sarcoméricos,
los mecanismos fisiopatológicos responsables de la hipertrofia, la evolución
natural y las estrategias de tratamiento no son los mismos.
– El criterio ecocardiográfico
de amiloidosis quedó definido por la presencia de HVI
con un punto de corte de ≥12 mm a nivel septal
o pared posterior según 10th Internacional Symposium on Amyloidosis 2004.
Score de Mayo predictor genotípico en MCH
|
Variable clínica |
Puntos |
|
Edad < 45 años |
1 |
|
Grosor de la pared del ventrículo izquierdo
> 20 mm |
1 |
|
Antecedentes familiares
de MCH |
1 |
|
Antecedentes familiares de muerte súbita
cardiaca |
1 |
|
Forma septal inversa |
1 |
|
Hipertensión arterial (HTA) |
-1 |
Los hallazgos de la NGS se compararon con el Mayo
Score (rango de -1 a 5) según las variables clínicas y ecocardiográficas.
Un paciente con la puntuación Mayo 5 tenía una mutación patogénica (100 % de
rendimiento). Los pacientes con una puntuación de Mayo de 4 tenían una mutación
patogénica en el 71 % de los casos. Los pacientes con una puntuación de Mayo de
3 o 2 tenían una mutación patogénica en el 50 y el 35 % de los casos,
respectivamente. El rendimiento de las pruebas genéticas con una puntuación de
-1 a 1 fue bajo (6-21%).
ANEXO
2
Maron et al 9 establecieron una clasificación morfológica
de cuatro tipos: tipo I, hipertrofia septal-anterior;
tipo II, hipertrofia septal-anterior y septal-posterior; tipo III, hipertrofia septal
y antero-lateral, y tipo IV, hipertrofia septal-posterior
y/o antero-lateral.
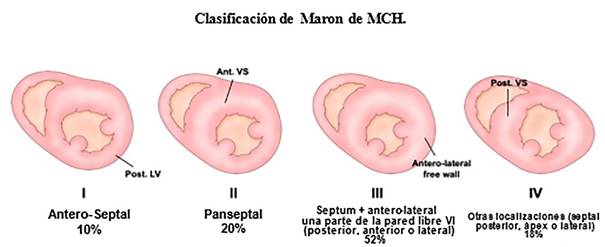
Modificado de: Maron BJ. Hypertrophic cardiomyopathy. Curr Probl Cardiol.
1993 Nov;18(11):639-704. doi: 10.1016/0146-
2806(93)90025-w. PMID: 7903919.
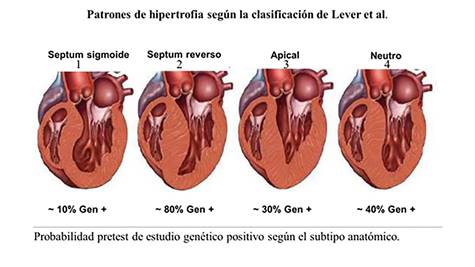
Esta clasificación ha resultado de gran utilidad a la
hora de valorar la probabilidad de presentar un estudio genético positivo
basado en el fenotipo anatómico expresado por el paciente, en lo que se ha
venido a llamar estudio genético guíado por
ecocardiografía.
Modificado de: Lever HM, Karam RF, Currie PJ, Healy BP. Hypertrophic
cardiomyopathy in the elderly. Distinctions from the young based on cardiac
shape. Circulation 1989 Mar; 79(3):580-9.67
ANEXO
3
Ejemplos de casos clinicos:
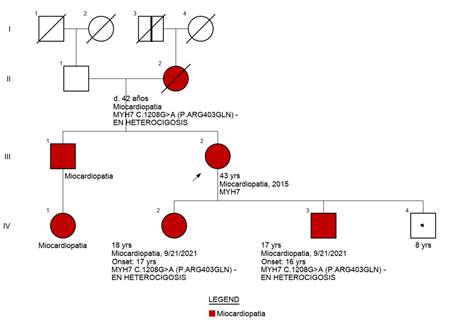
Ejemplo de Genograma de una familia evaluada.
Referencias: cuadrados: varones, círculos: mujeres, rojo: p con diagnóstico
clínico de MCH, blancos: p sin MCH ni mutación o no estudiado, símbolos con
punto negro central: portadores de mutación sin fenotipo de MCH, símbolos con
barra negra vertical: sujetos con posible MCH por historia clínica (no
comprobado). Línea diagonal: p fallecidos, flecha: caso índice.
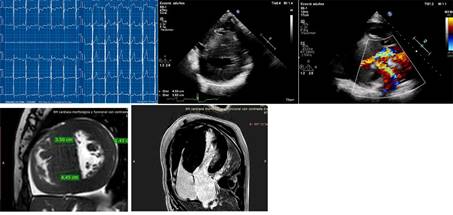
El caso índice es una mujer con hipertrofia severa
diagnosticada a los 37 años, con implante de CDI por taquicardia ventricular
(TV) monomorfa sostenida sincopal a los 47 años. La
CRM que evidenció Miocardio no compacto (MNC) y G+ (MYH7: c.1208G>A (p.
Arg403Gln). Sus 3 hijos presentan G+, dos de ellos con F+, ECG patológico
(hipertrofia ventricular izquierda y ondas T negativas en la cara anterolateral) ECG y CRM de uno de sus hijos arriba y de la
paciente abajo.
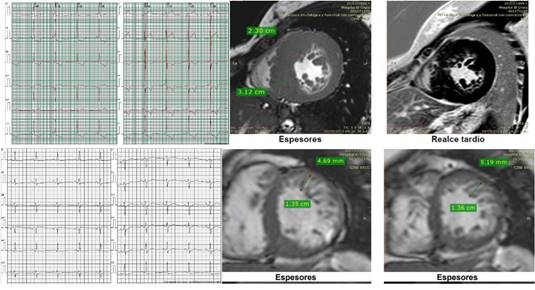
Caso 2. Adolescente de 17 años con angor
de esfuerzo (F+ y G-) con VUS+
ANEXO
4
Análisis
mutacional
A partir de muestras de saliva recogidas mediante un
hisopado bucal El ADN genómico obtenido de la muestra enviada se enriquece para
las regiones seleccionadas mediante un protocolo basado en la hibridación y se
secuencia mediante la tecnología Illumina. Todas las
regiones seleccionadas se secuencian con una profundidad ≥50x o se
complementan con análisis adicionales.
Las lecturas se alinean con una secuencia de
referencia (GRCh37) y los cambios de secuencia se identifican e interpretan en
el contexto de una sola transcripción clínicamente relevante, que se indica a
continuación.
El enriquecimiento y el análisis se centran en la
secuencia de codificación de los transcritos indicados, 20 pb
de secuencia intrónica flanqueante
y otras regiones genómicas específicas que han demostrado ser causantes de la
enfermedad en el momento del diseño del ensayo. Los promotores, las regiones no
traducidas y otras regiones no codificantes no se interrogan de otro modo. Para
algunos genes, solo se analizan los loci específicos
(indicados en la tabla anterior). Las eliminaciones y duplicaciones exónicas se llaman mediante un algoritmo interno que
determina el número de copias en cada objetivo al comparar la profundidad de
lectura para cada objetivo en la secuencia probando con la profundidad de
lectura media y la distribución de profundidad de lectura, obtenidas de un
conjunto de datos clínicos. Los marcadores en los cromosomas X e Y se analizan
con fines de control de calidad y pueden detectar desviaciones del complemento
de cromosomas sexuales esperado. Tales desviaciones pueden incluirse en el informe
de acuerdo con las directrices internas. La confirmación de la presencia y la
ubicación de las variantes notificables se realiza
según los criterios estrictos establecidos por Invitae
(1400 16th Street, San Francisco, CA 94103, n.° 05D2040778), según sea
necesario, utilizando uno de varios enfoques ortogonales validados (PubMed ID 30610921). Los siguientes análisis se realizan si
son relevantes para la solicitud. Para los exones 12 a 15 de PMS2, el genoma de
referencia se modificó para obligar a todas las lecturas de secuencias
derivadas de PMS2 y el pseudogen PMS2CL a alinearse
con PMS2, y los algoritmos de llamada de variantes se modificaron para admitir
una expectativa de 4 alelos. Si se identifica una variante rara de SNP o indel mediante este método, tanto PMS2 como el pseudogén PMS2CL se amplifican mediante PCR de largo
alcance y la ubicación de la variante se determina mediante la secuenciación
SMRT de Pacific Biosciences
(PacBio) del exón relevante en ambos amplicones de largo alcance. Si se identifica una CNV, se
ejecuta MLPA o MLPA-seq para confirmar la variante.
Si se confirma, tanto PMS2 como PMS2CL se amplifican mediante PCR de largo
alcance, y PacBio secuencia la identidad de las
diferencias fijas entre PMS2 y PMS2CL a partir del amplicón
de largo alcance para desambiguar la ubicación de la CNV.
El componente técnico de la secuenciación
confirmatoria lo realiza Invitae Corporation
(1400 16th Street, San Francisco, CA 94103, #05D2040778). Para la prueba de
expansión repetida de C9orf72, las unidades repetidas de hexanucleótidos
se detectan mediante PCR con cebado repetido (RP-PCR) con cebadores marcados
con fluorescencia, seguido de electroforesis capilar. Rangos de referencia de
interpretación: benigno (rango normal): <25 unidades repetidas, incierto:
25-30 unidades repetidas, patógeno (mutación completa): >=31 unidades
repetidas. Se usa una segunda ronda de RP-PCR que utiliza un conjunto de
cebadores que no se superponen para confirmar la llamada inicial en el caso de
tamaños de alelo sospechosos de 22 o más repeticiones. Para el análisis de ARN
de los genes indicados en la tabla Genes Analyzed, el
ADN complementario se sintetiza mediante transcripción inversa a partir de ARN
derivado de una muestra de sangre y se enriquece con secuencias de genes
específicas mediante hibridación de captura. Después de la secuenciación de
alto rendimiento con la tecnología Illumina, las
lecturas de salida se alinean con una secuencia de referencia (construcción del
genoma GRCh37; derivado personalizado del transcriptoma
RefSeq) para identificar las ubicaciones de las
uniones de exón a través de la detección de lecturas divididas. El uso relativo
de las uniones exónicas en una muestra de prueba se
evalúa cuantitativamente y se compara con el uso observado en las muestras de
control. El uso anormal de la unión exónica se evalúa
como evidencia en el marco de interpretación de variantes de Sherloc. Si se pronostica un patrón de empalme anormal en
base a una variante de ADN fuera del rango notificable
típico, como se describió anteriormente, la presencia de la variante se
confirma mediante secuenciación de ADN específica. La secuenciación del ARN la
realiza Invitae Corporation
(1400 16th Street, San Francisco, CA 94103, #05D2094793). Invitae
Corporation (5 Technology
Drive, Irvine CA 92618, n.° 05D1052995) realiza el componente técnico del
cultivo de células de fibroblastos y la extracción de gDNA
a partir de una biopsia con sacabocados de la piel.
Un PMID es un identificador único que hace referencia
a un artículo científico publicado. Busque por PMID en http://www.ncbi.nlm.nih.gov/pubmed.
Un rsID es un identificador
único que hace referencia a una sola posición genómica y se utiliza para
asociar información de frecuencia de población con cambios de secuencia en esa
posición. Las frecuencias de población informadas se derivan de varios sitios
públicos que agregan datos de proyectos de secuenciación de población a gran
escala, incluidos ExAC (http://exac.broadinstitute.org), gnomAD (http://gnomad.broadinstitute.org)
y dbSNP (http://ncbi.nlm.nih.gov/SNP).
Una ID de MedGen es un identificador
único que hace referencia a un artículo en MedGen, la
base de datos centralizada de información sobre trastornos genéticos y
fenotipos del NCBI.
Busque por ID de MedGen en http://www.ncbi.nlm.nih.gov/medgen.
Un número OMIM es un identificador único que se refiere a una entrada completa
en la herencia mendeliana en línea del hombre (OMIM). Busque por número OMIM en
http://omim.org/. Invitae
utiliza información de personas que se someten a pruebas para informar la
interpretación de variantes. Si se cita “Invitae”
como referencia en los detalles de la variante, esto puede referirse al
individuo en esta solicitud y/o a las observaciones internas históricas