
INTRODUCCIÓN
La miocardiopatía hipertrófica (MCH) es la cardiopatía genética más frecuente, con una prevalencia de hasta 1 en 500 personas. (1) Constituye una enfermedad compleja y variable en cuanto a fisiopatología, presentación clínica, pronóstico y sobrevida. Es la primera causa de muerte súbita en jóvenes y atletas; sin embargo, la mayoría de los pacientes cursan asintomáticos, lo que dificulta muchas veces el diagnóstico. La MCH genética habitualmente corresponde a una enfermedad de tipo monogénico con herencia autosómica dominante. (2) La mayoría de las variantes o mutaciones detectadas son en genes de proteínas que conforman el sarcómero. (3) Aunque el hallazgo de resultado positivo del estudio genético (EG) es variable según el fenotipo, llega a ser tan alto como 70-80 % en grupos seleccionados. (4) Identificar la base genética en la MCH no solamente permite realizar estudios diagnósticos en cascada a familiares, sino que también aporta valiosa información pronóstica. (5,6) No obstante, esta herramienta aún no se utiliza de manera rutinaria por su alto costo y difícil acceso. Definir el grupo de pacientes en quienes es más esperable un EG positivo puede ser de utilidad para abordar esta problemática.
OBJETIVOS
Describir las características clínicas y de estudios complementarios en una cohorte de pacientes con MCH en seguimiento en un centro no especializado, y analizar qué variables se asocian significativamente con un EG positivo.
MATERIAL Y MÉTODOS
Estudio de cohorte retrospectiva de pacientes evaluados en un centro no especializado desde enero de 2022 a enero de 2025, con diagnóstico de MCH por ecocardiograma Doppler color transtorácico (ETT), con al menos un segmento del ventrículo izquierdo (VI) con espesor parietal ≥15 mm, o ≥13 mm en familiares de pacientes con diagnóstico certero de MCH, en ausencia de hipertensión u otra enfermedad cardíaca, sistémica o congénita que la explique. (1) Se analizaron los datos clínicos, antecedentes familiares, electrocardiograma (ECG), ETT, estudios de esfuerzo, ECG Holter, resonancia magnética cardíaca (RMC) con gadolinio y el EG, cuando se encontrara disponible. Se excluyeron aquellos pacientes con fenocopias como amiloidosis o enfermedad de Fabry, y pacientes con otras causas de cardiopatía como valvulopatías graves.
Estudio genético
El EG se realizó con técnica de next generation sequencing (NGS), con base en un panel de miocardiopatías hipertróficas. El mismo debía incluir, como mínimo, un panel básico. Este panel contiene los genes prioritarios, es decir, aquellos con suficiente evidencia clínica y funcional para ser considerados asociados a la enfermedad, así como los genes relacionados con las principales fenocopias de esta patología. (1,7) Siguiendo esta línea, los paneles utilizados debían incluir el análisis de como mínimo 10 genes del sarcómero (MYH7, MYBPC3, TNNT2, TPM1, MYL2, MYL3, TNNI3, TNNC1, TPM1, ACTC1 y CSRP3), 4 genes de otras estructuras celulares del miocito (PTPN11, PLN, JPH2, DES) y 4 genes de patologías de almacenamiento (GLA, LAMP2, PRKAG2, TTR) para descartar fenocopias. El ácido desoxirribonucleico (ADN) fue extraído de manera automatizada de sangre entera en papel de filtro o de saliva. Se buscaron variantes de un solo nucleótido (SNV) y pequeñas inserciones y deleciones (indels) con una cobertura mínima de 20 fragmentos superior al 98 %. La clasificación de las variantes siguió las directivas del Colegio Americano de Genética Médica y Genómica 2015 (ACMG), (8) y se caracterizaron como patogénicas, probablemente patogénicas (probabilidad > 90 % de ser patogénica), de significado incierto (VUS de sus siglas en inglés), probablemente benignas (probabilidad > 90 % de ser benigna) o benignas.
Análisis estadístico
Las variables categóricas (presentadas como frecuencia y porcentaje) se compararon mediante prueba de Chi-cuadrado o test exacto de Fisher, según la frecuencia de los valores esperados. Las variables continuas de distribución normal se presentan como media y desviación estándar (DE) y las no gaussianas como mediana y rango intercuartílico (RIC). La evaluación de normalidad de la distribución se realizó mediante análisis de gráficos (histograma, plots) o el test de Shapiro Wilk. La comparación de las variables cuantitativas se realizó mediante el test de t o el Wilcoxon rank sum test según correspondiera. La asociación de las variables evaluadas con un estudio genético positivo se exploró en una tabla de contingencia. Todas las variables que alcanzaron un valor de p < 0,10 ,así como aquellas consideradas clínicamente relevantes, se incluyeron en un modelo de regresión logística múltiple, para definir las variables independientemente asociadas con un EG positivo. Se consideró significativo un valor de p < 0,05.
Consideraciones éticas
El protocolo del estudio fue revisado y aprobado por el Comité de ética de la Institución. La investigación fue conducida en concordancia con la declaración de Helsinki. (9) Todos los participantes del estudio otorgaron su consentimiento informado para participar del mismo.
RESULTADOS
Se incluyeron 72 pacientes, con edad promedio de 54 ± 16 años, 55 (76,4 %) de sexo masculino. Veintisiete pacientes (37,5 %) tenían antecedentes familiares asociados de MCH o muerte súbita. Veintinueve pacientes (40,3 %) tenían hipertensión arterial, en todos los casos leve y en tratamiento con un único fármaco. Referían algún síntoma 43 pacientes (59,7 %), el más frecuente la disnea. El 83,3 % de los pacientes presentaba cambios en el ECG, 41,7 % criterios de hipertrofia ventricular izquierda (HVI) y 61,1 % ondas T negativas. Presentaban obstrucción en el tracto de salida del ventrículo izquierdo (OTSVI) 28 pacientes (39,4 %), y en 4 (5,5 %) se había indicado una terapéutica de reducción septal. Portaban cardiodesfibrilador implantable (CDI), por prevención primaria o secundaria, 9 pacientes (12,5 %). Los patrones de HVI por ecocardiograma más comunes fueron el septum tipo reverso en 33 (45,8 %), septum sigmoideo en 29 (40,3 %) e hipertrofia apical en 8 (11,1 %). El promedio del máximo espesor parietal por ecocardiograma fue de 18,5 ± 3,9 mm. Se obtuvo información de la RMC en 61 de los 72 pacientes; 49 (81,7 %) presentaban algún grado de realce tardío con gadolinio (RTG), en su mayoría leve (<5 % de la masa miocárdica); solo 16 un RTG extenso (>15 % de la masa). El promedio del score de riesgo de muerte súbita de la Sociedad Europea (HCM Risk-SCD) (10) fue 2,7 ± 1,9 (riesgo < 4 % a 5 años). El resto de los parámetros se encuentran descritos en la Tabla 1.
Tabla 1
Características basales de la población (n=72)
| Variables | |
|---|---|
| Edad, años | 46,4 ±16,3 |
| Sexo Masculino | 55 (76,4) |
| Antecedentes familiares MCH | 27 (37,5) |
| Hipertensión arterial | 29 (40,3) |
| Fibrilación auricular | 19 (26,4) |
| Cardiodesfibrilador implantable | 9 (12,5) |
| Síntomas | 43 (59,7) |
| ECG | |
| Signos HVI | 30 (41,7) |
| Ondas T Negativas | 44 (61,1) |
| ETT | |
| Máximo espesor septal, mm | 18,57 ±3,9 |
| Septum reverso | 33 (45,8) |
| Septum sigmoideo | 29 (40,3) |
| HVI tipo apical | 8 (11,1) |
| Obstrucción dinámica VI | 28 (39,4) |
| Insuficiencia mitral ≥ leve | 45 (61,4) |
| Movimiento anterior sistólico, válvula mitra | 29 (41,4) |
| FEVI, % | 60 ± 11 |
| Diámetro aurícula izquierda, mm | 42,5 ± 6,6 |
| Disfunción diastólica ≥leve | 62 (85,7) |
| RMC | |
| Máximo espesor, mm | 19,1 ± 5,3 |
| Anomalía valva anterior mitral | 27 (45) |
| Inserción apical músculos papilares | 19 (33,3) |
| RTG Presente | 49 (81,7) |
| Score HCM Risk-SCD | 2,7 ± 1,9 |
ECG: electrocardiograma; ETT: ecocardiograma transtorácico; FEVI: fracción de eyección ventricular izquierda; HVI: hipertrofia ventricular izquierda; MCH: miocardiopatía hipertrófica; mm: milímetros; RMC: resonancia magnética cardíaca; RTG: realce tardío con gadolinio.VI: ventrículo izquierdo Las variables cualitativas se presentan como frecuencia y porcentaje, y las cuantitativas como media ± desviación estándar.
Si bien el EG fue indicado de rutina en todos los pacientes para evitar sesgos de selección, por cuestiones relacionadas con ausencia de cobertura, costo, o negativa del paciente, finalmente se llevó a cabo en 41 de los 72 pacientes (57 %). En la comparación entre estos 41 pacientes con los 31 restantes no se encontraron diferencias significativas en las variables de mayor relevancia clínicas, ecocardiográficas o de resonancia cardíaca.
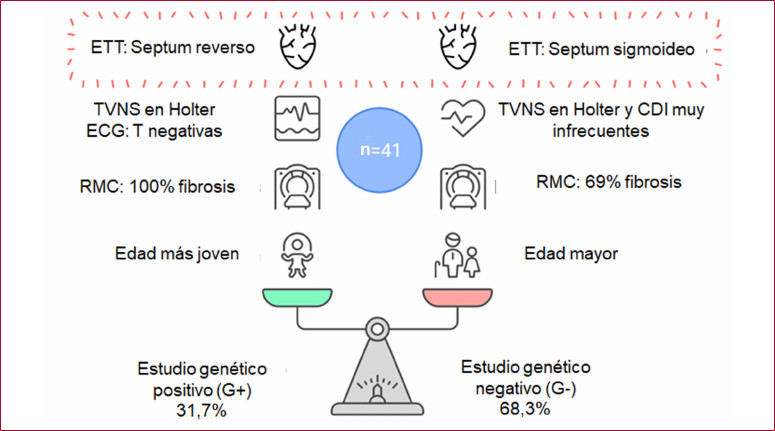
En 13 (31,7 %) de los paneles genéticos realizados se obtuvo un resultado positivo para una mutación sarcomérica (grupo G+): 5 en MYBPC3, 5 en MYH7, 2 en TNNT2 y 1 en FLNC. Los 28 casos restantes (68,3 %) fueron resultados negativos o VUS en genes que no se asocian fenotípicamente con MCH (grupo G-). Los pacientes G+ eran más jóvenes al momento del diagnóstico (edad media 38,4 ±15,3 vs. 50,8 ±11,7 años, p=0,007). El género y los factores de riesgo cardiovascular fueron similares en ambos grupos. No hubo diferencias en la presencia de síntomas, considerando disnea, angina, palpitaciones o síncope. En el ECG se observaron con mayor frecuencia ondas T negativas en los pacientes G+ (92,3 % vs 42,9 %, p=0,003), así como mayor frecuencia de taquicardia ventricular no sostenida (TVNS) en el ECG Holter (61,5 % vs 10,5 %, p=0,002). En los estudios por imágenes, los pacientes G+ presentaban más frecuentemente fenotipo de HVI tipo septum reverso (84,6 % vs 17,9 %), y los G- con mayor frecuencia fenotipo de septum sigmoideo (67,9 % vs. 7,7 %), en ambos casos con diferencia estadísticamente significativa. Asimismo, los pacientes G+ presentaban en el ETT mayor espesor parietal máximo, con mediana (RIC) de 21 (17,7-22,5) mm en G+ vs. 17 (16-18,8) mm en G-, p=0,016; y menor diámetro auricular izquierdo: 38,6 ±5,8 mm vs 44,5 ±4,5 mm, p=0,001. Se obtuvo información de la RMC con gadolinio en 37 de los 41 pacientes. Todos los pacientes G+ presentaron algún grado de RTG, frente al 69,6 % de los 28 pacientes G- (p=0,027).
Con respecto al riesgo de muerte súbita, los pacientes G+ presentaban mayor valor del score europeo HCM Risk-SCD, con una mediana de 2,3 (1,8-4,5) vs 1,8 (1,4-2,7), p=0,038. Seis de los 13 pacientes G+ ya tenían, o les fue implantado durante el seguimiento, un CDI, vs solo 1 de los 28 pacientes G- (p<0,001). Los resultados de la comparación entre los pacientes G+ y los G- se encuentran en la Tabla 2. En el análisis multivariado únicamente la presencia de septum de tipo reverso se asoció de manera independiente a un resultado positivo en el estudio genético (OR 21,5, IC 95 % 2,72-171,28, p=0,004).
Tabla 2
Hallazgos clínicos y de estudios complementarios según la presencia de estudio genético positivo (G+) o negativo (G-).
| Variables | G+ (n=13) | G- (n=28) | p |
|---|---|---|---|
| Edad, años | 45,3 ±16,4 | 57,1 ±11,7 | 0,012 |
| Edad diagnóstico, años | 38,4 ±15,3 | 50,8 ±11,7 | 0,007 |
| Sexo masculino | 12 (92,3) | 21 (75%) | 0,056 |
| Antecedentes familiares MCH/MS | 8 (61,5) | 6 (21,4) | 0,012 |
| Hipertensión arterial | 6 (46,2) | 21 (75) | 0,072 |
| Síntomas | 8 (61,5) | 20 (71,4) | 0,527 |
| Cardiodesfibrilador implantable | 6 (46,2) | 1 (3,6) | 0,001 |
| ECG/Holter | |||
| Ondas T negativas | 12 (92,3) | 12 (42,9) | 0,003 |
| Signos HVI | 7 (53,8) | 9 (32,1) | 0,185 |
| TVNS | 8 (61,5) | 2 (10,5) | 0,002 |
| ETT | |||
| Septum reverso | 11 (84,6) | 5 (17,9) | 0,001 |
| Septum sigmoideo | 1 (7,7) | 19 (67,9) | 0,001 |
| Máximo espesor septal, mm | 21 (17,7-22,5) | 17 (16-18,8) | 0,016 |
| FEVI, % | 60 (56-66) | 63 (51-65) | 0,958 |
| Diámetro AI, mm | 38,6 ± 5,84 | 44,5 ± 4,52 | 0,001 |
| Obstrucción dinámica | 3 (23,1) | 11 (39,3) | 0,308 |
| Insuficiencia mitral ≥ leve | 7 (53,8) | 16 (57,1) | 0,467 |
| Movimiento anterior sistólico válvula mitral | 5 (38,5) | 9 (32,1) | 0,750 |
| Disfunción diastólica ≥ leve | 12 (92,3) | 23 (82,1) | 0,504 |
| RMC | |||
| RTG presente | 13 (100) | 16 (69,9) | 0,027 |
| Máximo espesor septal, mm | 19,6 (17,8-22,8) | 17,4 (14,7-20,1) | 0,049 |
| Inserción apical músculos papilares | 5 (41,7) | 5 (23,8) | 0,283 |
| Score HCM Risk-SCD | 2,3 (1,8-4,5) | 1,8 (1,4-2,7) | 0,038 |
AI: aurícula izquierda; ECG: electrocardiograma; ETT: ecocardiograma transtorácico; FEVI: fracción de eyección ventricular izquierda; HVI: hipertrofia ventricular izquierda; MCH: miocardiopatía hipertrófica; mm: milímetros; MS: muerte súbita; RMC: resonancia magnética cardíaca; RTG: realce tardío con gadolinio; TVNS: taquicardia ventricular no sostenida; VI: ventrículo izquierdo
Las variables cualitativas se presentan como frecuencia y porcentaje, y las cuantitativas como media ± desviación estándar. o mediana y rango intercuartílico
La Figura 1 resume los hallazgos más relevantes del análisis.
DISCUSIÓN
Correlación genotipo-fenotipo
La MCH se expresa con un fenotipo variable, y la correlación genotipo-fenotipo es motivo de continua investigación. En nuestro estudio encontramos que el 31,7 % de la población genotipificada presentó una variante patogénica (o probablemente patogénica) en proteínas del sarcómero. Las dos variantes más frecuentemente encontradas fueron en el gen que codifica a la proteína C cardíaca de unión a la miosina (MYBPC3) y en el de la cadena pesada de la beta-miosina (MYH7), similar a lo descripto en la literatura. (1)
La característica que más fuertemente se asoció a un EG positivo fue la presencia de un septum tipo reverso. Desde las primeras descripciones de correlación genotipo-fenotipo, como la del grupo de la Mayo Clinic, se ha demostrado que esta morfología es el predictor más fuerte de un EG positivo, con hasta 79 % de G+ vs 8 % en pacientes con septum sigmoideo. (11) Nuestros resultados fueron similares: el 68,8 % de los pacientes con septum tipo reverso fueron G+ vs solo el 5 % de los pacientes con septum sigmoideo. En análisis bayesiano, la presencia de septum reverso elevó la probabilidad de un test genético positivo desde un valor pre test del 31,7 % hasta un 71 %, con una razón de verosimilitud positiva de 5,17. Esto refuerza que el rédito del estudio en pacientes seleccionados puede ser verdaderamente alto.
En la cohorte general, el hallazgo de EG positivo fue similar al 38 % reportado por la Mayo Clinic en una población no seleccionada. El score de Mayo es una herramienta de predicción de resultado positivo del EG, que incluye la edad de diagnóstico <45 años, máximo espesor septal ≥20mm, antecedente familiar de MCH o de muerte súbita, y septum tipo reverso en el ecocardiograma (un punto por cada factor), y la presencia concomitante de hipertensión arterial como factor predictor negativo (-1 punto). (12) Este score presenta múltiples validaciones posteriores, con correcta especificidad, pero variable sensibilidad. (13,14) Un estudio reciente en Argentina mostró un rédito del 80 % en pacientes con score de Mayo ≥3. (15) En nuestro estudio, al igual que en el score, además del septum tipo reverso previamente mencionado, los pacientes G+ tenían menor edad al momento del diagnóstico y mayor carga de antecedentes familiares, y presentaban mayor espesor parietal máximo que los pacientes G-, aunque estas variables no lograron significancia en el análisis multivariado, posiblemente por el escaso número de pacientes.
Implicancias pronósticas
El 46,2 % de los pacientes G+ tenía o le fue colocado un CDI durante el seguimiento en el consultorio, comparado con solo el 3,6 % de los pacientes G-. Además, el grupo G+ presentó mayor prevalencia de TVNS en el ECG Holter y mayor valor del score de riesgo de muerte súbita (HCM Risk-SCD), en concordancia con la bibliografía. (16) Múltiples estudios han demostrado que un estudio genético positivo representa mayor riesgo de muerte, disfunción ventricular, insuficiencia cardíaca y necesidad de trasplante. (17,18) Incluso hay evidencia de variantes de mayor riesgo como las mutaciones en MYH7, principalmente en regiones altamente conservadas como la región conversora 19. No obstante, cuando se ajusta por otros factores, la asociación entre variantes sarcoméricas y muerte súbita pierde significancia, (20) por lo que las guías aún no colocan el genotipo como determinante para la colocación de un CDI. (1,21)
Finalmente, el hallazgo de fibrosis en la resonancia cardíaca en nuestro estudio fue mucho más frecuente en el grupo G+. No obstante, pese a asociarse claramente a peor pronóstico, se sabe que algún grado de RTG es un hallazgo muy frecuente en los pacientes con MCH, (22) por lo que esta característica no permite discriminar correctamente pacientes de mayor riesgo.
Perspectivas clínicas y aplicabilidad al mundo real
Actualmente las guías de manejo de miocardiopatías de 2023 y 2024 recomiendan realizar EG en pacientes con MCH (recomendación grado I, nivel de evidencia B) en los casos en que permite confirmar el diagnóstico, evaluar el pronóstico, estratificar el tratamiento y dar consejo reproductivo. (1,21) No obstante, en Argentina el acceso a estudios genéticos es limitado (23) y depende muchas veces del costo directo para el paciente. En este contexto, identificar predictores fenotípicos que aumenten la probabilidad pretest de EG positivo puede ser útil para justificar la indicación ante financiadores o aseguradoras, lo cual puede tener un impacto directo en facilitar el acceso.
Nuestro estudio valida que algunos patrones fenotípicos (como el septum reverso) pueden aumentar significativamente el rédito del EG. Además, el presente estudio fue llevado a cabo en un centro no especializado en cardiopatías familiares, lo cual refuerza su aplicabilidad clínica en el contexto del mundo real, donde la mayoría de los pacientes son atendidos en centros generales.
LIMITACIONES
La limitación más relevante de nuestro estudio es que si bien la prueba genética fue indicada en todos los pacientes para evitar sesgos de selección, y que representa a una cohorte general, sólo se pudo obtener el resultado en poco más del 55 % de los casos, principalmente por el alto costo del estudio y la ausencia de cobertura aún por el sistema de salud. Un registro multicéntrico de Argentina recientemente publicado, que incluyó datos de 8 provincias obtuvo un valor incluso menor de estudios genéticos realizados, (23) lo cual refuerza la realidad de la dificultad del acceso al mismo en todo el país. Por otro lado, en la mayoría de los casos el panel de NGS realizado fue básico, e incluía únicamente los genes prioritarios y principales fenocopias. Si bien esta práctica se encuentra respaldada por la bibliografía, posiblemente realizando un panel ampliado de genes el rendimiento del estudio puede aumentar. (1) Ambas limitaciones son reflejo del alto costo de esta herramienta y su difícil acceso. Es claro que la introducción de una medicina de precisión y un enfoque basado en la genética benefician al paciente, pero también es cierto que permanentemente afrontamos las limitaciones que imponen la situación socioeconómica y las restricciones de las distintas coberturas de salud. Finalmente cabe resaltar que, si bien el análisis multivariado arrojó una asociación estadísticamente significativa entre el septum tipo reverso y el EG positivo, el intervalo de confianza extremadamente amplio refleja una elevada incertidumbre en la estimación, claramente relacionada con el tamaño de la muestra. Por esta razón, decidimos no reportar este hallazgo como un factor asociado de manera concluyente y su resultado debe ser tomado con cautela.
CONCLUSIÓN
En una cohorte de pacientes con MCH, un tercio de aquellos en quienes se realizó EG presentaron mutaciones en proteínas del sarcómero. La presencia de un septum de tipo reverso parece ser la variable más fuertemente asociada a un EG positivo. Dadas las limitaciones en nuestro medio para acceder al estudio en pacientes con miocardiopatías, conocer las variables que podrían asociarse con un mayor rédito diagnóstico, puede contribuir a refinar la selección de los pacientes candidatos a esta herramienta.
Declaración de conflicto de intereses
Los autores declaran que no tienen conflicto de intereses.
(Véanse formularios de conflicto de intereses de los autores en la Web).
Fuente de apoyo:
El presente trabajo no recibió ninguna beca o financiación.